Epigenetic Regulation
Background
Epigenetic regulation describes mechanisms to control gene expression without altering the genetic information encoded in the DNA. Such regulation can be achieved via chemical modifications of the DNA and the associated histone proteins. In particular, the positioning and modifications of nucleosomes define active or repressive chromatin domains and demarcate regulatory elements. This organization is crucial for the dynamic regulation of gene expression in differentiation, development and response to external stimuli. In addition, many diseases like cancer rely on alterations of chromatin features. Hence it is essential to understand the dynamics of chromatin state transitions and their consequences for gene expression. Post-translational modifications (PTM) of histones and the regulatory roles of chromatin-associated RNAs are in the focus of our research. We address these questions at specific model loci or on a genome-wide scale in the context of specific external stimuli or diseases like Leukemia.
Work of the group
Mechanisms of chromatin regulation by RNA
RNA is an integral component of chromatin, and recent findings demonstrate its essential role in shaping the structure and dynamics of the epigenome. Specific functions of chromatin-associated RNAs (caRNAs) include recruitment and modulation of epigenetic modifiers and transcription factors, as well as the modulation of enhancer-promoter interactions, among others. In order to fully understand how the epigenome regulates gene expression in health and disease, the contribution of RNA in shaping chromatin states needs to be dissected in more detail. However, targeted perturbation of these molecules in their native environment remains challenging and there is an unmet need for technologies that enable manipulation of RNA binding at endogenous gene loci. We address this question by developing strategies for site-directed RNA recruitment in mammalian cells and apply those to study functional roles of caRNAs.
Chromatin states and transcriptional dynamics
Transcription is a dynamically regulated process regarding both the input from active transcription factors binding to the promoter or enhancer and the production of RNA over time. We study the light induced transcription of a model gene to infer the transitions of promoter states in gene activation, silencing and re-activation. Fluorescence microscopy and reversible light-induced recruitment of transcription factors to our gene of interest allow us to analyze transcriptional time courses with defined start times in single cells and to deduce the underlying mechanisms by quantitative modelling. In addition, we employ perturbations of chromatin modifications, e.g. of histone acetylation, to identify chromatin marks that characterize distinct promoter states.
Cause and consequence of chromatin alterations on IFNβ-induced gene expression
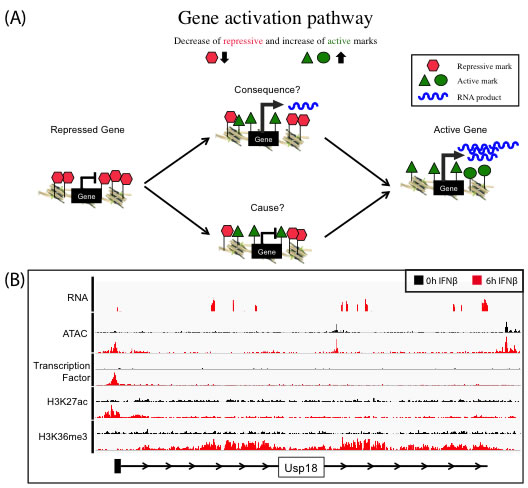
We use the well-characterized JAK-STAT pathway as a model to understand the induction of gene expression by an external stimulus, i.e., the cytokine interferon beta (IFNβ). We investigate the epigenetic changes of target promoter sites and regulatory regions like enhancers upon IFNβ stimulus by using multiple genomic approaches including RNA-seq, ChIP-seq and ATAC-seq. The characterization of the epigenome on multiple levels and over time allow conclusions about cause and consequence of chromatin alterations on gene expression (Figure 1A). The transcriptional induction (RNA-seq) is captured and linked to the dynamics in histone modification patterns and transcription factor binding (ChIP-seq). Gene activation can lead to the local reorganization of nucleosomes, creating an area of increased chromatin accessibility that can be measured by ATAC-seq (Figure 1B).
|
Figure 1. (A): Concept of gene activation pathway to understand the function of histone marks as cause or consequence of transcription. Genes marked by repressive histone marks (red) do not show transcription, in contrast to active gene, which do express RNA (blue) and are marked with several active histone marks (green). The exact dynamics of removing repressive and setting active marks to allow gene activation process are not fully understood, especially what is cause and consequence is unknown. (B) Representative browser tracks of various Next Generation Sequencing techniques (RNA-Seq, ATAC-Seq, ChIP-Seq). Gene activation of target gene (Usp18) in embryonic mouse stem cells treated for 6h (red) with IFNβ in contrast to 0h control (black)(RNA-Seq). Enrichment of all marks at the transcription start site (ATAC-Seq, transcription factor ChIP-Seq, H3K27ac ChIP-Seq) and in the gene body (H3K36me3 ChIP-Seq). |
Selected references
Pankert T, Jegou T, Caudron-Herger M & Rippe K (2017). Tethering RNA to chromatin for fluorescence microscopy based analysis of nuclear organization. Methods 123, 98-101. doi: 10.1016/j.ymeth.2017.01.010 | Abstract | Reprint (4.2 MB).
Molitor J, Mallm JP, Rippe K & Erdel F (2017). Retrieving chromatin patterns from deep sequencing data with correlation functions. Biophys J 112, 473-490. doi: 10.1016/j.bpj.2017.01.001 |Abstract | Reprint (10.3 MB) | Software | Article metrics.
Müller-Ott K, Erdel F, Matveeva A, Hahn M, Mallm, JP, Rademacher A, Marth C, Zhang Q, Kaltofen S, Schotta G, Höfer T & Rippe K (2014). Specificity, propagation and memory of pericentric heterochromatin. Mol Syst Biol 10, 746. doi: 10.15252/msb.20145377 | Abstract | Reprint (7.4 MB) | Article metrics
Teif VB, Beshnova DA, Vainshtein Y, Marth C, Mallm JP, Höfer T & Rippe K (2014). Nucleosome repositioning links DNA (de)methylation and differential CTCF binding during stem cell development. Genome Res 24, 1285-1295. doi: 10.1101/gr.164418.113 | Abstract | Reprint (3.3 MB) | Article metrics